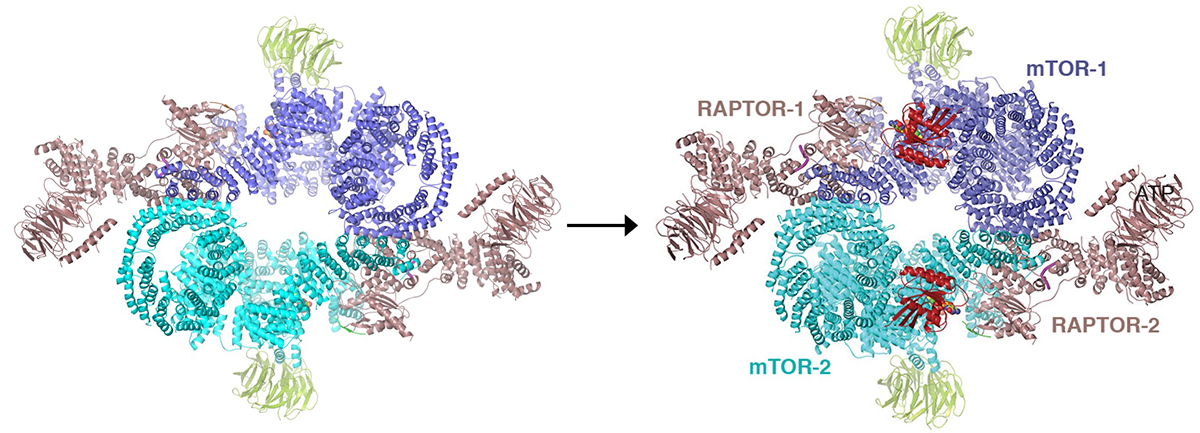
The mammalian target of rapamycin (mTOR) protein, a member of the phosphoinositide 3-kinase (PI3K)-related protein kinase (PIKK) family, has a central role in two growth-regulatory pathways that have distinct inputs and downstream effectors. In one pathway, mTOR assembles with its RAPTOR subunit to form mTOR Complex 1 (mTORC1), while in the other pathway RAPTOR is replaced by RICTOR to form mTORC2. mTORC1 activation requires nutrients, which are sensed as amino acid levels and induce mTORC1 recruitment to lysozomal membranes through RAPTOR. There, mTORC1 meets its activator, the small GTPase RHEB, which conveys a second set of signals from environmental cues including energy, oxygen levels, and growth factors. In metazoan, growth factors are also needed to relieve the inhibition of mTORC1 by PRAS40. In earlier work, we had determined the crystal structure of truncated mTOR containing the kinase domain and the associated FAT domain shared with all PIKKs. This established the PIKK kinase catalytic mechanism and also revealed that the binding site for the immunosuppressant/antiproliferative agent rapamycin is a highly conserved site adjacent to the catalytic cleft, raising the question of whether it is a substrate-recruitment domain (Yang et al., 2013). This we confirmed with a crystal structure of the rapamycin-binding domain bound to a substrate-peptide.
In subsequent work, we used cryo-EM to determine the 3.1 Å structure of the intact mTORC1 complex, as well as the 3.4 Å structure of activated mTORC1-RHEB complex (Yang et al., 2017). We found that RHEB binds 60 Å away from the kinase active site, yet allosterically rearranges active site residues by ~3 Å, causing a ~30-fold increase in kcat. The activation mechanism pointed to the FAT domain being an auto-inhibitory domain, a function likely shared by other PIKKs. The autoinhibitory function is consistent with cancer-associated hyperactivating mutations mapping either to the FAT domain, the FAT-kinase interface, or the interface between the two kinase lobes. This suggested that the mutations lower the barrier to the kinase adopting the active configuration, essentially mimicking the effects of RHEB. We confirmed this hypothesis by showing that the mutations lower the EC50 of activation by RHEB and that they do not synergize with saturating RHEB concentrations.