Wild-type RAS GTPases (H, N and KRAS) cycle between an active, GTP–bound, and an inactive, GDP–bound, state. KRAS mutations, particularly KRASG12C, are one of the most frequent activating mutations in lung cancer, a disease responsible for 150,000 annual deaths in the US. Cancer-causing mutations impair the GTPase activity of KRAS, causing it to accumulate in the activated state. Unlike KRASWT, which cycles between its active and inactive forms, KRAS oncoproteins are thought to be ‘constitutively’ active in cancer cells. Despite the prevalence of these mutations, no therapies that directly target this oncoprotein are currently available in the clinic. A recently identified binding pocket in KRASG12C has enabled the discovery of compounds that potently inhibit KRAS-GTP or effector signaling by this mutant.
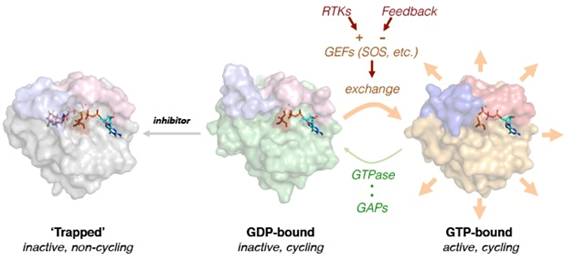
In recent work we described the mechanism by which novel KRASG12C inhibitors suppress KRASG12C-signaling and cancer cell proliferation (Science, 2016; PMID: 26841430). Inhibition requires that KRASG12C has basal GTPase activity in cancer cells and occurs because drug-bound KRASG12C is insusceptible to nucleotide exchange factors, and thus trapped in its inactive state. Indeed, mutants completely lacking GTPase activity and those promoting exchange reduced the potency of the drug. Suppressing nucleotide exchange activity downstream of various tyrosine kinases enhanced KRASG12C inhibition, whereas its potentiation had the opposite effect. These findings reveal that KRASG12C undergoes nucleotide cycling in cancer cells and provide a basis for developing effective therapies to treat KRASG12C-driven cancers.
Current efforts are directed at identifying other modulators of response to these inhibitors in lung cancer and determining how mutant KRAS drives the proliferation of cancer cells.